Introduction
MyPROSLE implements a new methodology to summarize the transcriptome of individual patients in a series of biological functions or gene-modules, which were later used to predict up to a total of 111 different clinical outcomes, including clinical manifestations, laboratory measurements, cytokine levels, autoantibodies, cell percentages, nearby disease flares and response to drugs.
To understand a little better how everything works, let's see a brief introduction:
First key point is that the method should reflect the molecular portrait of the patients, for this we decided to use gene expression data because we consider that transcriptome is a good reflection of what is happening in a patient at the molecular level. But transcriptome contains thousands and thousands of genes, and so it is not easily interpretable. In fact, genes do not act alone, rather form regulatory networks acting in specific biological functions. Therefore, we decided to project the genes into 600 gene modules, or networks, previously described, related with immune functions. In this way, we reduce the dimensionality from thousands of genes to hundreds of gene-modules.
Now, we need to be able to quantify how deregulated each gene-module is in each patient with respect to healthy controls. For this, we create M-scores (formula 1). Briefly, the M-score of a gene-module in a patient is calculated by adding the z-score of all its genes with respect to healthy controls. That is, the further the value is from 0, the more dysregulated the gene module is with respect to the healthy state, both positive and negative.
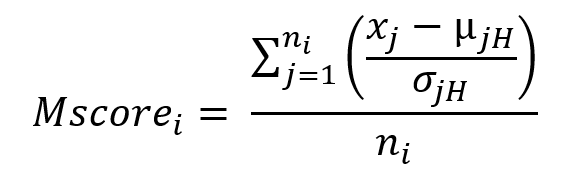
Where xj is the expression of gene j in an individual patient, μxjH and σxjH are the mean of the expression and the standard deviation of gene j in healthy samples and n is the number of genes from the module i. The M-scores fit a centered normal distribution. By definition, a value of 1.65 and -1.65 in such distribution will cover 95 percent of the positive and negative values, respectively, which would correspond to a p-value of 0.05 for each tail. Therefore, we can consider statistically significant those M-scores that take values greater than 1.65 (in absolute value).
At this point, we had about 600 gene-modules, but not all of them have to be relevant to SLE. Therefore, we selected only the gene-modules that are significantly dysregulated in at least 10% of patients in at least 3 datasets. In this way, we selected gene-modules that may be relevant in small groups but that are consistent across dataset. 206 gene-modules were selected.
Finally, we performed a clustering to see how the different gene-modules are grouped, and we obtained 9 very clear clusters of gene-modules that summarized 9 main biological functions related with SLE, or SLE-signatures (Figure 1).
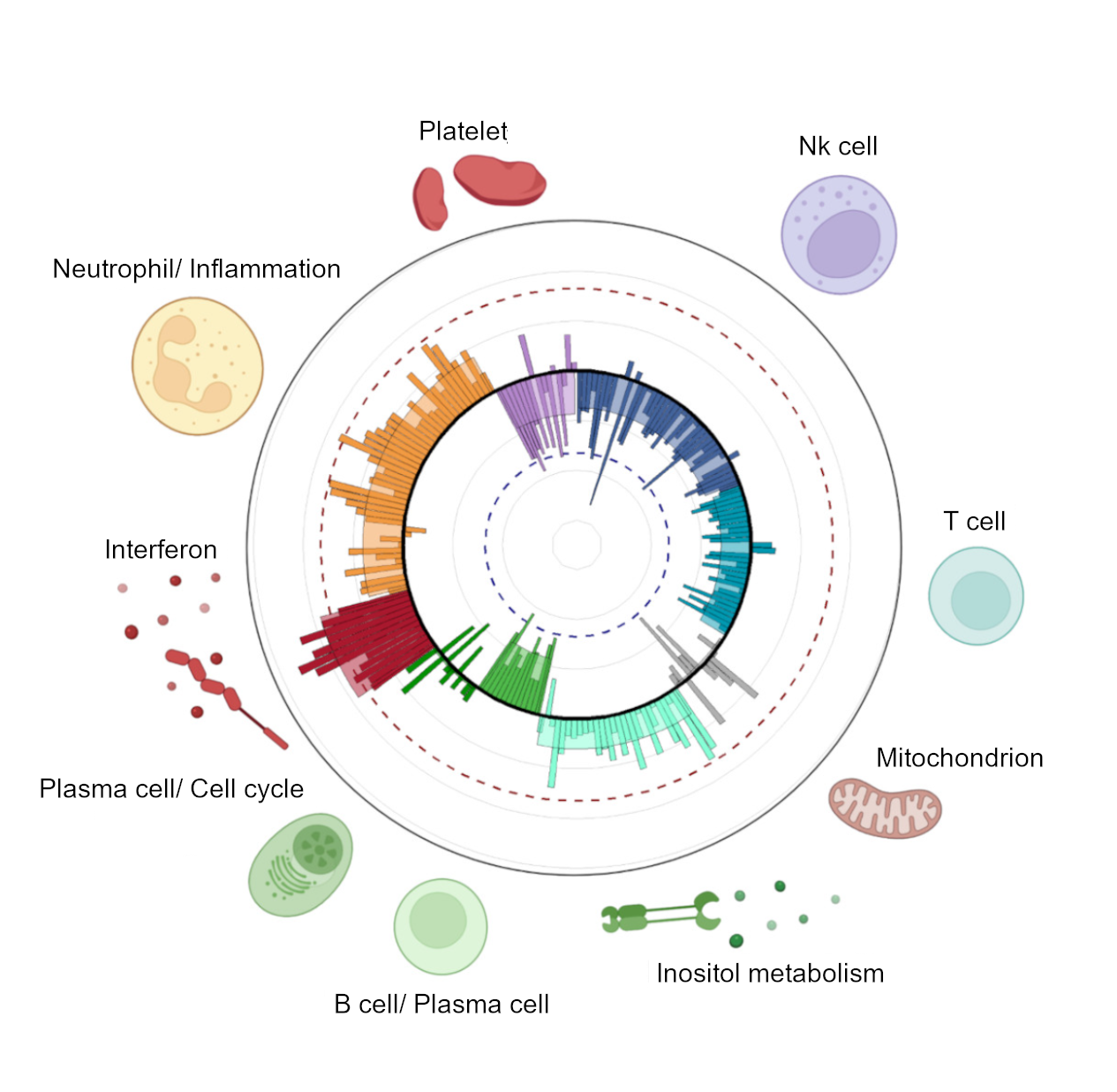
As conclusion of this part: We can summarize the molecular portrait of a patient into 206 functional gene-modules or in 9 main SLE-signatures. And the M-scores of each module/signature quantifies how dysregulated that biological pathway is with respect to the healthy state.
The imputation of the M-scores for a new patient without providing control samples is carried out as follows: 1) the expression of the patient is centered and scaled in the same way as the expression of the samples contained in our internal database of SLE patients, 2) Euclidean distance between the expression profile of the new patient and each patient in our database is estimated, 3) the k most similar patients from our database to the new patient are then selected, and 4) M-scores for the new patient are imputed as the mean of the M-scores of the k most similar patients.
Next, we use the values of M-scores of a total of more than 6000 patients to build prediction models for different clinical outcomes, which can be applied to any new patient through this same website, for this, we are going to see the section How to use.
How to use
First, go to the Analysis page. MyPROSLE works with patient expression data (from 1 patient), preferably already normalized and transformed to logarithm. You can upload your data in a tabulated table where the first column would be the genes (in Gene Symbol) and the following columns would correspond to the different patients. You can see the format in more detail in Figure 2 or download an example.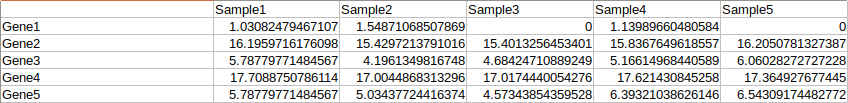
M-scores can be calculated using healthy controls as reference or by patient-patient similarity. If you do not enter data from healthy individuals, the second methodology is selected by default.
To test, we can click on the Run an example button (found in the Analysis section) and data for 15 Lupus patients will be automatically loaded.
Once the expression data of the patients (and healthy controls, optionally) have been selected/loaded, we click on the Run Analysis button.
First, MyPROSLE calculates the M-scores for each patient and for each of the 206 Gene-modules. At this point, we obtain a figure like figure 1 personalized for each patient, as well as a table with all the M-scores per patient and per gene-modules and also with the median M-scores based on the SLE-signature at which that each Gene-module belongs to (Section Personalized molecular profiling).
If healthy samples are not supplied, the mean distance between each sample and the most similar reference patients is reported in the first row of the M-scores table. It should be checked by the authors and, if necessary, samples with large distance (e.g., >30) should be discarded from the analysis. It may happen that all samples have a large distance, in which case that dataset is not appropiate to be used without matched healthy controls.
Once we have the M-scores for a patient, MyPROSLE applies the prediction models previously generated for different clinical outcomes to it. In section Predict clinical outcomes, the tables with the predictions for each clinical variable per patient can be viewed and downloaded. You can download the following table to obtain detailed information about how to interpret the results for each clinical variable, as well as to see the performance results of each of the applied models.
Download supplementary tableAttention
The previous table reports all results from the classification, both variables with good and poor prediction results. Therefore, this should be carefully taken into account to interpret the predictions performed on your data. For instance, proliferative nephritis is predicted accurately in our test dataset (AUC=0.98, Sensitivity=0.83, Specificity=0.94) and, therefore, this variable is expected to be predicted accurately. However, other variables have a worse performance in the test data. For instance, myositis have an specificity of 0 and, therefore, the predictions for this variable are not trustworthy. we emphasize that users should use metrics such as balanced accuracy, since this metric is more robust against possible imbalances in the data. Similarly, we advise paying close attention to the sensitivity and specificity of the different predictive models to ensure that the model predicts both classes well. For quantitative variables, users must assess the models based on the correlation and Rsquared values they present. We are constantly working on improving our models incorporating new data and algorithms.
MyPROSLE was designed for research purposes, should not be used in the clinical practice.
Contact us if you have any doubt or suggestion.
Updates
- 2023-09: Euclidean distance between samples are now displayed if healthy controls are not provided.
Citation
If you use MyPROSLE, please include the following citation:
Toro-Domínguez et al, Scoring personalized molecular portraits identify Systemic Lupus Erythematosus subtypes and predict individualized drug responses, symptomatology and disease progression, Briefings in Bioinformatics, Volume 23, Issue 5, 2022, https://doi.org/10.1093/bib/bbac332
Team
Daniel Toro-Domínguez 1Contact: daniel.toro@genyo.es
Marta E. Alarcón-Riquelme 1,2Pedro Carmona-Sáez 1,3
Manuel Martínez-Bueno 1
Raúl López-Domínguez 1,3
Jordi Martorell-Marugán 1,3
Elena Carnero-Montoro 1
Guillermo Barturen 1
Daniel Goldman 4
Michelle Petri 4
1 GENYO. Centre for Genomics and Oncological Research: Pfizer, University of Granada, Andalusian Regional Government
2 Karolinska Institute
3 University of Granada
4 John Hopkins University School of Medicine







Load data
Personalized molecular profiling
Figure 1. Molecular portrait of the patient. Each bar represents a gene module and its M-score colored by SLE signature. SLE-signatures summarize the main biological functions involved in the development of the pathology.
Predict clinical outcomes
Figure 2. Clinical manifestations predicted as positive or present in the patient. The clinical manifestations were grouped by categories according to the main tissue or organ they affect. The color of each category (represented by an affected organ icon) depends on the number or percentage of positive clinical manifestations within each category. Review the right table or Figure 3 for more details on the predicted clinical manifestations in each case.
Figure 3. The figure shows in the x-axis the absence/Presence probability (from 0 to 1) of suffering each clinical manifestation for categorical variables, or the predicted value for numerical variables (in picogram/milliliter for cytokines and in percentage for cell types) for each patient. Descriptions of each clinical variable and predictive model performances can be downloaded from help tab.
Supported by:

